SGTPy.component
SGTPy.component object stores pure component information needed to evaluate SAFT-VR-Mie EoS for phase equilibria and interfacial properties computation.
The simplest scenario is coarse-grained (CG) non-associating non-polar fluids. The parameters needed for these molecules are the numbers of segments (\(m_s\)), well-depth of Mie potential (\(\epsilon\)) in K units, size parameter of Mie potential (\(\sigma\)) in Å (\(10^{-10}\) m), attractive (\(\lambda_a\)) and repulsive (\(\lambda_r\)) exponents of Mie Potential A component can be created as follows:
>>> from SGTPy import component
>>> methane = component('methane', ms=1.0, sigma=3.752 , eps=170.75, lambda_r=16.39, lambda_a=6.)
>>> dodecane = component('dodecane', ms=4.0, sigma=4.351, eps=378.56, lambda_r=18.41, lambda_a=6.)
The molecular parameters for CG molecules can be obtained from the Bottled-saft webpage.
Additionally, a simple corresponding state parametrization [1] is available through SGTPy. This parametrization requires the critical temperature (Tc), acentric factor (w) and depend on the desired number of segments (\(m_s\)) and the liquid density at a reduced temperature equal to 0.7 in mol/m \(^3\).
>>> hexane = component('hexane', Tc= 507.6, w=0.301261)
>>> hexane.saftvrmie_forcefield(ms=2, rhol07=6973.5)
>>> # sigma in meters, epsilon in K and lambda_r
... (4.510744361846902e-10, 373.5197182392722, 19.43607049398115)
The SAFT-VR-Mie EoS has been extended to homonuclear rings [2], for these type of fluids and additional geometric parameter (\(\chi\), ring in SGTPY) is needed.
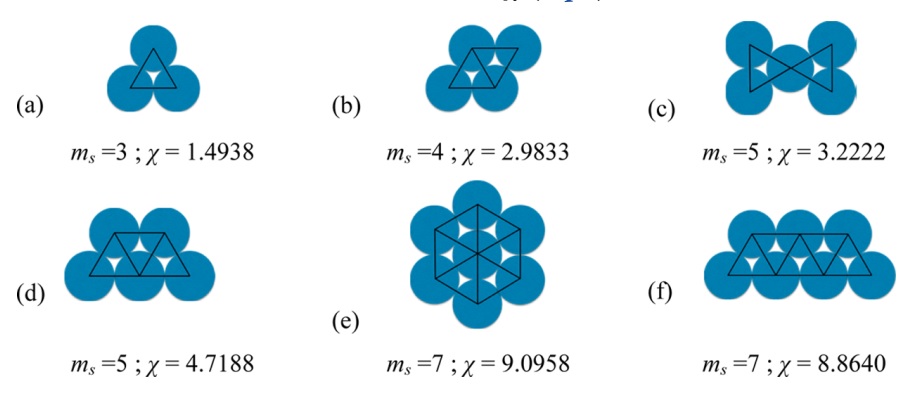
A ring molecule can be created as follows:
>>> benzene = component('benzene', ms=3, sigma=3.441, eps=230.30, lambda_r=10.45, lambda_a=6.,
... ring=1.4938)
For the case of pure self-associating fluid, three extra parameters are needed: the association energy (\(\epsilon^{AB}\)) in K units, the association range (\(r_c^{AB}/\sigma\)) and association center position (\(r_d^{AB}/\sigma\)). the association scheme is characterized by the triple [B, P, N], which indicates the number of bipolar, positive and negative association sites, respectively. An equivalence table for common association schemes and the [B, P, N] triplet is shown below.
Association Scheme |
[B, P, N] |
|---|---|
1 |
[0, 0, 1] |
2B |
[0, 1, 1] |
2C |
[1, 0, 1] |
3B |
[0, 1, 2] |
4B |
[0, 3, 1] |
4C |
[0, 2, 2] |
Parameters for self-associating fluid, i.e. alcohols and water, for SAFT-VR-Mie can be found in Ref. [3]. An associating molecule can be created as follows:
>>> water = component('water', ms = 1.7311, sigma = 2.4539 , eps = 110.85,
... lambda_r = 8.308, lambda_a = 6., eAB = 1991.07, rcAB = 0.5624,
... rdAB = 0.4, sites = [0,2,2])
Additionally, a polar contribution [4] can be accounted for, this contribution requires the definition of a dipolar moment (\(\mu\), mupol in SGTPy) in Debye units, and the number of polar sites (\(n_p\), npol in SGTPy). Molecular parameters for polar molecules can be found in Ref. [3] and [5].
>>> butanol = component('butanol2C', ms = 1.9651, sigma = 4.1077 , eps = 277.892,
... lambda_r = 10.6689, lambda_a = 6., eAB = 3300.0, rcAB = 0.2615,
... rdAB = 0.4, sites = [1,0,1], npol = 1.45, mupol = 1.6609)
Finally, in order to model the interfacial behavior, the influence parameter (\(c_{ii}\)) in J m \(^5\) / mol \(^2\) is required.
>>> water = component('water', ms = 1.7311, sigma = 2.4539 , eps = 110.85,
... lambda_r = 8.308, lambda_a = 6., eAB = 1991.07, rcAB = 0.5624,
... rdAB = 0.4, sites = [0,2,2], cii = 1.5371939421515458e-20)
>>> butanol = component('butanol2C', ms = 1.9651, sigma = 4.1077 , eps = 277.892,
... lambda_r = 10.6689, lambda_a = 6., eAB = 3300.0, rcAB = 0.2615,
... rdAB = 0.4, sites = [1,0,1], npol = 1.45, mupol = 1.6609,
... cii = 1.5018715324070352e-19)
Warning
User is required to supply the necessary parameters for the EoS to work properly